Пмл 1100 характеристики: Магнитный пускатель ПМЛ-1100 220В в Брянске. Цена, описание, характеристики, купить
alexxlab | 31.01.2023 | 0 | Разное
Магнитный пускатель ПМЛ 1100 нереверсивный на 10А Этал
12 товаров
Сортировать по
АкцииОт дешевых к дорогимОт дорогих к дешевымПопулярныеРейтингНовинкиПо количеству просмотровПо названию (А-Я)
Магнитный пускатель ПМЛ 1100 Украинского производителя Этал, применение, расшифровка маркировки, технические характеристики, принцип действия, размеры и информация по доставке и отправке.
На нашем сайте Вы можете купить пускатель ПМЛ 1100 от официального дистрибьютора Этал, цены и остатки на продукцию обновляются каждый день полностью полагайтесь на сайт, отправка товара производится по всей Украине.
Пускатель ПМЛ 1100 применяется для пуска прямым подключением и остановки трехфазных асинхронных электрических двигателей категории АС-3 с короткозамкнутым ротором.
Для категории АС-3 мощность двигателя при напряжении:
220В – 2,2 кВт;
380В – 4 кВт
500В – 5,5 кВт.
Расшифровка маркировки серии пускателей ПМЛ 1100:
ПМЛ 1100 О*4 220В 10А IP00: ПМЛ – наименование серии; “1″ первой величины на номинальный ток 10 Ампер; “1” указывает на отсутствие теплового реле; нереверсивный магнитный пускатель; “0” – степень пыле-влаго защиты по ГОСТ 14254-96; “0” – на переменный ток с одним размыкающим контактом; “О” – климатическое исполнение; “четыре” согласно ГОСТ 15150-69; класс износостойкости А 3,0 млн. циклов; номинальное напряжение катушки 220 Вольт.
Для избегания ряда неточностей рекомендуем уточнить нужный вам параметр из списка напряжений втягивающей катушки: 24, 36, 40, 48, 110, 127, 220, 380, 660 Вольт с частотой 50 Герц.
Технические характеристики ПМЛ 1100:Номинальное напряжение пускателей составляет 660 Вольт;
Номинальный ток 10 Ампер;
Степень защиты IP00 (без оболочки)
Без встроенного теплового реле
Во время номинальной работы втягивающая катушка пускателя потребляет мощность 8 ± 1,8 В·А, а при запуске
мощность увеличивается до 68 ± 8 В·А.
При категории АС-3 механическая и коммутационная износостойкость аппарата изменяется в зависимости от исполнения А, Б и В.
Класс А – составляет до 20 миллионов циклов механической и до 3 миллионов циклов коммутационной износостойкости;
Класс Б – составляет до 10 миллионов циклов механической и до 1,5 миллионов циклов коммутационной износостойкости;
Класс В – составляет до 3 миллионов циклов механической и до 300 тысяч циклов коммутационной износостойкости.
Категорией применения АС-3 предполагается прямой пуск и отключение двигателя без преждевременной остановки асинхронных двигателей с коротко замкнутым ротором вращающихся без остановки. Категория применения АС-3 допускает не долгое торможение противотоком двигателя или незапланированное повторно-кратковременное включение, но при таких режимах работы количество коммутаций не должно превышать пяти в течении одной минуты и десяти в течении 10 минут.
Принцип действия магнитного пускателя ПМЛ 1100:Для того чтобы запустить электродвигатель нужно кратковременно зажать кнопку «Пуск» – вследствие этого по катушке пускателя начнет проходить электрический ток, потом произойдет притягивание якоря к сердечнику, а главные контакты пускателя которые присоединены к цепи питания замкнутся. В месте с этими процессами в сцепление вступит вспомогательный контакт. После возврата кнопки «Пуск» в исходное положение ток не прекратит протекать по катушке.
В месте с этими процессами в сцепление вступит вспомогательный контакт. После возврата кнопки «Пуск» в исходное положение ток не прекратит протекать по катушке.
Описанный выше принцип называется «самоблокировка» он гарантирует электрическому двигателю нулевой степень защиты, при таком режиме работы в случаях чрезмерного падения напряжения, магнитное поле катушки ослабевает, а вспомогательные контакты расходятся. Для повторного запуска электродвигателя нужно опять зажать кнопку «Пуск». Если напряжение отсутствовало, а потом появилось, степень защиты IP00 блокирует незапланированный запуск электродвигателя. Из-за данного качества пускатели применяются для управления станками на предприятиях. Для отключения двигателя необходимо нажать на кнопку «Стоп» это приведёт к размыканию главных контактов, а также к угасанию магнитного поля.
Габаритные и установочные размеры ПМЛ 1100, его вес:
Пускатель крепится с применением двух винтов М4 или на монтажную рейку.
Габаритные размеры – Ш/73,8хД/67хВ/44мм
Установочные размеры – 50х35 мм
Электрическая схема:
Контакторы ПМЛ производятся согласно ГОСТ 50030.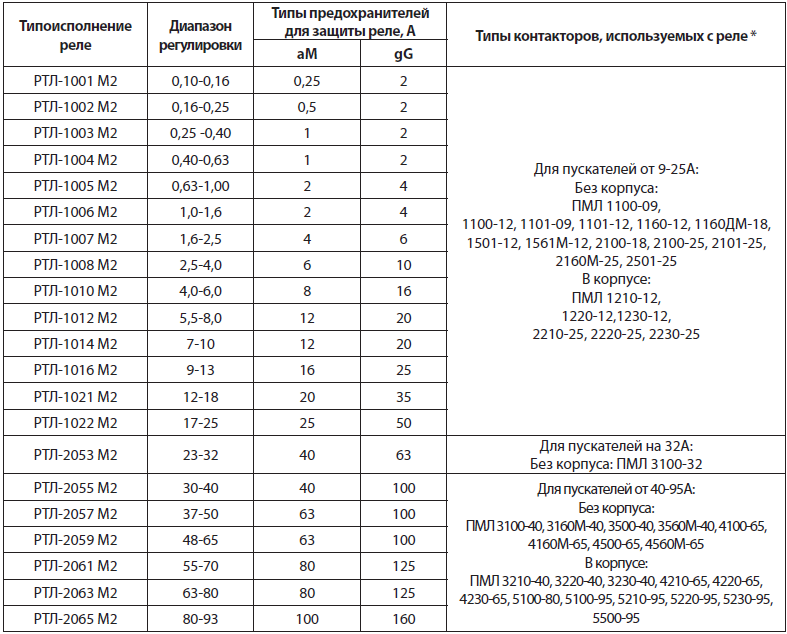 4.1 и ГОСТ 2491-82
4.1 и ГОСТ 2491-82
Заменяют малогабаритные контакторы серии КМИ 10910 IEK. Самостоятельно подобрать замену, Вы можете с помощью электронной таблицы в формате Word.
На сайте указаны актуальные цены и остатки на ПМЛ 1100, на складе в городе Харьков поддерживаются часто запрашиваемые позиции пускателей и реле, при этом ассортимент постоянно обновляется.
Электромагнитный пускатель ПМЛ – Низкая цена
Расшифровка маркировки
ПМЛ – ХХХХ
ПМЛ Название серии пускателя
Условное обозначение номинального тока главных контактов пускателя:
первая величина — 10А, при наличии буквы Д -16А
вторая величина — 25А
третья величина – 32, 40А
четвертая величина- 63А, при наличии в обозначении буквы Д 80А
пятая величина — 125А
шестая велична — 160А
седьмая величина — 250А
Условное обозначеие исполнения пускателей по назначению и наличию теплового реле:
1 – пускатель нереверсивный, тепловое реле РТЛ в комплект не входит
2 – пускатель нереверсивный с тепловым реле РТЛ
5 – реверсивный пускатель без теплового реле РТЛ с механической блокировкой для пускателей со степенью защиты ІР00 и ІР20. С электрической и механической блокировкой для пускателей степени защиты ІР40 и ІР54
С электрической и механической блокировкой для пускателей степени защиты ІР40 и ІР54
6 – реверсивный пускатель с тепловым реле РТЛ с электрической и механической блокировками в комплекте
7 – пускатель со схемой включения «звезда-треугольник».
Обозначение пускателя по степени защиты и наличия кнопок и сигнальных ламп.
0 – степень защиты ІР00;
1 – степень защиты ІР54 без кнопок на защитном корпусе (для пускателей без теплового реле) или с кнопкой “Реле” (для пускателей ПМЛ с тепловым реле РТЛ)
2 – степень защиты ІР54 с кнопками управления Пуск и Стоп на защитном корпусе
3 – степень защиты ІР54 с кнопками управления Пуск Стоп и сигнальным ламповым индикатором;
4 – степень защиты корпуса ІР40 без кнопок управления
6 – степень защиты ІР20. (Сальники на контактных зажимах)
Описание
Магнитный пускатель ПМЛ – коммутационный аппарат для удаленного управления электромагнитными устройствами. Пускатель ПМЛ серии работает в цепях с напряжением 660В и частотой 50 и 60Гц.
Применение
- для пуска и остановки электродвигателей;
- в комплектации с тепловым реле для предотвращения перегрузок.
Монтаж: винтовой.
Малогабаритный прибор (размеры незначительно варьируются), может быть открытым или в корпусе из пластика или металла.
При выборе основную информацию о технических параметрах можно получить из обозначений:
- номинальный ток;
- тип и наличие реле;
- степень защиты и элементы управления;
Например, магнитный пускатель ПМЛ 1100 по первой цифре – 10А, максимальное значение возможно при маркировке 6 – 160А. Смотрим вторую “1”: нереверсивный, тепловое реле отсутствует. Третья величина указывает на оптимальные условия среды для установки и на присутствие кнопок. В рассматриваемом варианте “0” – IP00 (без корпуса, только для размещения в зданиях, без доступа солнечных лучей, влажности, пыли), кнопок нет.
Наш интернет-магазин предлагает ассортимент всех типов серии ПМЛ – оцените характеристики и закажите подходящий магнитный пускатель ПМЛ. Цена, выгодно отличающаяся от имеющихся на рынке, удобная доставка и гарантия производителя до 2 лет – вот несколько наших главных преимуществ. Если Вам необходима консультация – свяжитесь с нашими специалистами по тел.: +7 (343) 288-71-80 и получите ответы на любые вопросы.
Цена, выгодно отличающаяся от имеющихся на рынке, удобная доставка и гарантия производителя до 2 лет – вот несколько наших главных преимуществ. Если Вам необходима консультация – свяжитесь с нашими специалистами по тел.: +7 (343) 288-71-80 и получите ответы на любые вопросы.
Купить магнитный пускатель ПМЛ в Екатеринбурге Вы сможете с бесплатной доставкой! Стоимость отправки в другие города России уточняется в момент подтверждения заказа с клиентом.
Таблица рекомендуемой замены пускателей ПМЕ, ПМ12, ПМА, ПМЛ, КМИ
| соответствие пускателей серии ПМЛ к пускателям серии ПМ12 | ||||
| ПМЛ-1100 | ПМ12-010100 (1з) | ПМЛ-1110 | ПМ12-010110 (1з) | |
| ПМЛ-1101 | ПМ12-010101 (1р) | ПМЛ-1210 | ПМ12-010210 (1з) | |
| ПМЛ-1501 | ПМ12-010501 (2р) | ПМЛ-1220 | ПМ12-010220 (1з) | |
| ПМЛ-1160М | ПМ12-010150 (1з) | ПМЛ-1230 | ПМ12-010230 (1з) | |
| ПМЛ-1161М | ПМ12-010151 (1р | ПМЛ-1511 | ПМ12-010510 (4з+2р) | |
| ПМЛ-1561М | ПМ12-010551 (2р) | ПМЛ-1611 | ПМ12-010610 (4з+2р) | |
| ПМЛ-1140 | ПМ12-010140 (1з) | ПМЛ-1621 | ПМ12-010620 (4з+2р) | |
| ПМЛ-1541 | ПМ12-010540 (4з+2р) | ПМЛ-1631 | ПМ12-010630 (4з+2р) | |
| ПМЛ-2100 | ПМ12-025100 | (1з) | ПМЛ-2110 | ПМ12-025110 (1з) |
| ПМЛ-2101 | ПМ12-025101 | (1р) | ПМЛ-2210 | ПМ12-025210 (1з) |
| ПМЛ-2501 | ПМ12-025501 | (2р) | ПМЛ-2220 | ПМ12-025220 (1з) |
| ПМЛ-2160М | ПМ12-025150 | (1з) | ПМЛ-2230 | ПМ12-025230 (1з) |
| ПМЛ-2161М | ПМ12-025151 (1р) | ПМЛ-2511 | ПМ12-025511 (2з+4р) | |
| ПМЛ-2561М | ПМ12-025551 (2р) | ПМЛ-2611 | ПМ12-025611 (2з+4р) | |
| ПМЛ-2140 | ПМ12-025140 (1з) | ПМЛ-2621 | ПМ12-025621 (2з+4р) | |
| ПМЛ-2541 | ПМ12-025541 (2з+4р) | ПМЛ-2631 | ПМ12-025631 (2з+4р) | |
| ПМЛ-3100 | ПМ12-040150 (2з+1р) | ПМЛ-3210 | ПМ12-040210 (2з+1р) | |
| ПМЛ-3500 | ПМ12-040550 (4з+2р) | ПМЛ-3220 | ПМ12-040220 (2з+1р) | |
| ПМЛ-3160М | ПМ12-040150 (2з+1р) | ПМЛ-3230 | ПМ12-040230 (2з+1р) | |
| ПМЛ-3560М | ПМ12-040550 (4з+2р) | ПМЛ-3510 | ПМ12-040510 (4з+2р) | |
| ПМЛ-3140 | ПМ12-040140 (2з+1р) | ПМЛ-3610 | ПМ12-040610 (4з+2р) | |
| ПМЛ-3540 | ПМ12-040540 (4з+2р) | ПМЛ-3620 | ПМ12-040620 (4з+2р) | |
| ПМЛ-3110 | ПМ12-040110 (2з+1р) | ПМЛ-3630 | ПМ12-040630 (4з+2р) | |
| ПМЛ-4100 | ПМ12-063151 (2з+2р) | ПМЛ-4210 | ПМ12-063211 (2з+2р) | |
| ПМЛ-4500 | ПМ12-063551 (2з+2р) | ПМЛ-4220 | ПМ12-063221 (2з+2р) | |
| ПМЛ-4160М | ПМ12-063151 (2з+2р) | ПМЛ-4230 | ПМ12-063231 (2з+2р) | |
| ПМЛ-4560М | ПМ12-063551 (2з+2р) | ПМЛ-4510 | ПМ12-063511 (2з+2р) | |
| ПМЛ-4140 | ПМ12-063141 (2з+2р) | ПМЛ-4610 | ПМ12-063611 (2з+2р) | |
| ПМЛ-4540 | ПМ12-063541 (2з+2р) | ПМЛ-4620 | ПМ12-063621 (2з+2р) | |
| ПМЛ-4110 | ПМ12-063111 (2з+2р) | ПМЛ-4630 | ПМ12-063631 (2з+2р) | |
| ПМЛ-5100 | ПМ12-100150 (2з+2р) | ПМЛ-5500 | ПМ12-100500 (4з+4р) | |
| ПМЛ-5101 | ПМЛ-5501 | |||
| ПМЛ-5110 | ПМ12-100110 (2з+2р) | ПМЛ-5510 | ПМ12-100510 (4з+4р) | |
| ПМЛ-5111 | ПМЛ-5511 | |||
| ПМЛ-5210 | ПМ12-100210 (2з+2р) | ПМЛ-5610 | ПМ12-100610 (4з+4р) | |
| ПМЛ-5211 | ПМЛ-5611 | |||
| ПМЛ-6100 | ПМ12-160150 (2з+2р) | ПМЛ-6500 | ПМ12-160500 (4з+4р) | |
| ПМЛ-6101 | ПМЛ-6501 | |||
| ПМЛ-6110 | ПМ12-160110 (2з+2р) | ПМЛ-6510 | ПМ12-160510 (4з+4р) | |
| ПМЛ-6111 | ПМЛ-6511 | |||
| ПМЛ-6210 | ПМ12-160210 (2з+2р) | ПМЛ-6610 | ПМ12-160610 (4з+4р) | |
| ПМЛ-6211 | ПМЛ-6611 | |||
Технические характеристики и габаритные размеры.
 Рис1
Рис1Рис.2
Рис.3
Рис.4
| Наименование | ПМЛ1100 (рис.1) | ПМЛ2100 (рис.2) | ПМЛ3100 (рис.3) | ПМЛ4100 (рис.3) | ПМЛ5100 (рис.3) | ПМЛ6100 (рис.4) | ПМЛ7100 (рис.4) |
Номинальный ток, А | 10 | 25, 32 | 40 | 63 | 80,95 | 160 | 250 |
Амах | 47 | 59 | 79 | 79 | 87 | 171 | 204 |
Смах | 82 | 102 | 116 | 116 | 127 | 181 | 210 |
С1 | 115 | 135 | 149 | 149 | 160 | Приставки не применяются | Приставки не применяются |
С2 | 134 | 154 | 168 | 168 | 179 | Приставки не применяются | Приставки не применяются |
Вес, кг | 0,36 | 0,55 | 1,23 | 1,23 | 1,45 |
Наименование | Амах | Bмах | B1мах | Сmax | G | L | Q | Q1 | S | Ф | F | P |
| ПМЛ6100 (рис4) | 171 | 176 | 137 | 181 | 80±0. | 114 | 30 | 60 | 20 | М6х20 | 133 | 40 |
| ПМЛ7100 (рис4) | 204 | 205 | 145 | 210 | 96±0.1 | 143 | 39 | 66.5 | 25 | М10х30 | 147 | 48 |
 8
8Clinical Trials and Observations: Прогрессирующая мультифокальная лейкоэнцефалопатия после терапии ритуксимабом у ВИЧ-отрицательных пациентов: отчет о 57 случаях из проекта «Исследования нежелательных явлений и отчетов о лекарственных препаратах»
1. Astrom KE, Mancall EL, Richardson EP., Jr Прогрессирующая мультифокальная лейкоэнцефалопатия: ранее не распознанное осложнение хронического лимфатического лейкоза и болезни Ходжкина. Мозг. 1958; 81: 93–111. [PubMed] [Google Scholar]
Astrom KE, Mancall EL, Richardson EP., Jr Прогрессирующая мультифокальная лейкоэнцефалопатия: ранее не распознанное осложнение хронического лимфатического лейкоза и болезни Ходжкина. Мозг. 1958; 81: 93–111. [PubMed] [Google Scholar]
2. Major EO, Amemiya K, Tornatore CS, Houff SA, Berger JR. Патогенез и молекулярная биология прогрессирующей мультифокальной лейкоэнцефалопатии, демиелинизирующего заболевания головного мозга, вызванного вирусом JC. Клин Микробиол Ред. 1992;5:49–73. [Бесплатная статья PMC] [PubMed] [Google Scholar]
3. Koralnik IJ, Schellingerhout D, Frosch MP. История болезни Массачусетской больницы общего профиля. Еженедельные клинико-патологические упражнения (случай 14-2004): 66-летний мужчина с прогрессирующим неврологическим дефицитом. N Engl J Med. 2004; 350:1882–1893. [PubMed] [Google Scholar]
4. Gonzalez H, Bolgert F, Camporo P, Leblond V. Прогрессирующий мультифокальный лейкоэнцефалит (PML) у 3 пациентов, получавших стандартную дозу флударабина (FAMP). Гематол Клетка Ther. 1999;41:183–186. [PubMed] [Google Scholar]
Гематол Клетка Ther. 1999;41:183–186. [PubMed] [Google Scholar]
5. Power C, Gladden JG, Halliday W, et al. Ассоциация ПМЛ, связанная со СПИДом и не связанная со СПИДом, с отчетливым полиморфизмом p53. Неврология. 2000;54:743–746. [PubMed] [Google Scholar]
6. Кляйншмидт-ДеМастерс Б.К., Тайлер К.Л. Прогрессирующая многоочаговая лейкоэнцефалопатия, осложняющая лечение рассеянного склероза натализумабом и интерфероном бета-1а. N Engl J Med. 2005; 353:369–374. [PubMed] [Google Scholar]
7. Лангер-Гулд А., Атлас С.В., Грин А.Дж., Боллен А.В., Пеллетье Д. Прогрессирующая многоочаговая лейкоэнцефалопатия у пациента, получавшего натализумаб. N Engl J Med. 2005; 353:375–381. [PubMed] [Академия Google]
8. Van Assche G, Van Ranst M, Sciot R, et al. Прогрессирующая многоочаговая лейкоэнцефалопатия после терапии натализумабом болезни Крона. N Engl J Med. 2005; 353:362–368. [PubMed] [Google Scholar]
9. Biogen. [По состоянию на 2 марта 2009 г.]; обновление Tysabri. 2009 г., февраль; http://library.corporate-ir.net/library/14/148/148682/items/325387/50A3DDB4-521D–4A7A-AA57-5B35B92DD671_biibTysabri20Feb09.pdf.
2009 г., февраль; http://library.corporate-ir.net/library/14/148/148682/items/325387/50A3DDB4-521D–4A7A-AA57-5B35B92DD671_biibTysabri20Feb09.pdf.
10. Хартунг Х.П. Новые случаи прогрессирующей мультифокальной лейкоэнцефалопатии после лечения натализумабом. Ланцет Нейрол. 2009 г.;8:28–31. [PubMed] [Google Scholar]
11. Aksoy S, Harputluoglu H, Kilickap S, et al. Ритуксимаб-ассоциированные вирусные инфекции у больных лимфомой. Лейк-лимфома. 2007;48:1307–1312. [PubMed] [Google Scholar]
12. FDA. Предупреждение FDA: информация для медицинских работников. Роквилл, Мэриленд: 2006. Декабрь [По состоянию на 2 марта 2009 г.]. Ритуксимаб. http://www.fda.gov/cder/drug/InfoSheets/HCP/rituximab.pdf. [Google Scholar]
13. EMEA. Европейский отчет об общественной оценке: Mabthera (редакция) 2007 г. [По состоянию на 2 марта 2009 г.]; http://www.emea.europa.eu/humandocs/PDFs/EPAR/Mabthera/H-165-PI-en.pdf.
14. ВОЗ. Информационный бюллетень ВОЗ по фармацевтическим препаратам. [По состоянию на 2 марта 2009 г.]; 2007 г. http://www.who.int/medicines/publications/newsletter/PN_3_2007.pdf.
[По состоянию на 2 марта 2009 г.]; 2007 г. http://www.who.int/medicines/publications/newsletter/PN_3_2007.pdf.
15. Южный Сан-Франциско, Калифорния: Genentech; 2006. Ритуксимаб (Ритуксан) [вкладыш]. [Google Scholar]
16. Южный Сан-Франциско, Калифорния: Genentech; 2008. Ритуксимаб (Ритуксан) [вкладыш]. [Google Scholar]
17. Genentech и Biogen. Важное предупреждение о наркотиках Новая информация о безопасности. [По состоянию на 2 марта 2009 г.]; 2008 г. http://www.fda.gov/medwAtch/safety/2008/rituxan_DHCP_Final%209411700.pdf.
18. Беринг Ю.М., Вивес К., Банных С. Прогрессирующая многоочаговая лейкоэнцефалопатия у больного с В-клеточной лимфомой маргинальной зоны. Дж. Нейроонкол. 2007; 85: 289–290. [PubMed] [Google Scholar]
19. Brito-Babapulle F, Moule S, Stewart W, et al. Прогрессирующая многоочаговая лейкоэнцефалопатия (ПМЛ) после химиотерапии комбинацией ритуксимаба: ежегодная конференция Британского общества гематологии. Бр Дж Гематол. 2006; 133:36. [Академия Google]
[Академия Google]
20. Darbesio A, Bertoldo E, Guana R, Geda C. Прогрессирующая мультифокальная лейкоэнцефалопатия, осложняющая фолликулярную лимфому: возможная роль химиотерапии и лечения анти-CD20. 4-я Национальная конференция медицинской онкологии Аннотация. Энн Онкол. 2002;13:94. [Google Scholar]
21. Фрейм Валь С.Г., Фолвик М.Р., Торп С.Х. Прогрессирующая мультифокальная лейкоэнцефалопатия у больного лимфомой с полной ремиссией после лечения цитостатиками и ритуксимабом: отчет о клиническом случае и обзор литературы. Клин Нейропатол. 2007; 26: 68–73. [PubMed] [Академия Google]
22. Goldberg SL, Pecora AL, Alter RS, et al. Необычные вирусные инфекции (прогрессирующая многоочаговая лейкоэнцефалопатия и цитомегаловирусная болезнь) после высокодозной химиотерапии с аутологичным спасением стволовых клеток крови и перитрансплантационным ритуксимабом. Кровь. 2002; 99: 1486–1488. [PubMed] [Google Scholar]
23. Harris HE. Прогрессирующая многоочаговая лейкоэнцефалопатия у пациента с системной красной волчанкой, получавшего ритуксимаб. Ревматология (Oxf) 2008; 47: 224–225. [PubMed] [Академия Google]
Ревматология (Oxf) 2008; 47: 224–225. [PubMed] [Академия Google]
24. Хасан М.М., Тейлор П. Прогрессирующая многоочаговая лейкоэнцефалопатия при хроническом лимфоцитарном лейкозе. Бр Дж Гематол. 2005; 130:808. [PubMed] [Google Scholar]
25. Краник С.М., Моури Э.М., Розенфельд М.Р. Прогрессирующая мультифокальная лейкоэнцефалопатия после ритуксимаба в случае неходжкинской лимфомы. Неврология. 2007; 69: 704–706. [PubMed] [Google Scholar]
26. Ng C, Slavin MA, Seymour JF. Прогрессирующая многоочаговая лейкоэнцефалопатия, осложняющая макроглобулинемию Вальденстрема. Лейк-лимфома. 2003;44:1819–1821. [PubMed] [Google Scholar]
27. Pelosini M, Focosi D, Rita F, et al. Прогрессирующая мультифокальная лейкоэнцефалопатия: отчет о 3 случаях у ВИЧ-отрицательных гематологических пациентов и обзор литературы. Энн Хематол. 2007; 87: 405–412. [PubMed] [Google Scholar]
28. Rey J, Belmecheri N, Bouayed N, et al. Паповавирусная лейкоэнцефалопатия JC после лечения первой линии CHOP и ритуксимабом. Гематология. 2007;92:e101. [PubMed] [Google Scholar]
Гематология. 2007;92:e101. [PubMed] [Google Scholar]
29. Steurer M, Clausen J, Gotwald T, et al. Прогрессирующая мультифокальная лейкоэнцефалопатия после аллогенной трансплантации стволовых клеток и посттрансплантационного ритуксимаба. Трансплантация. 2003; 76: 435–436. [PubMed] [Академия Google]
30. Yokoyama H, Watanabe T, Maruyama D, Kim SW, Kobayashi Y, Tobinai K. Прогрессирующая многоочаговая лейкоэнцефалопатия у пациента с B-клеточной лимфомой во время химиотерапии, содержащей ритуксимаб: клинический случай и обзор литературы. Int J Гематол. 2008; 88: 443–447. [PubMed] [Google Scholar]
31. Hopfinger G, Plessl A, Grisold W, et al. Прогрессирующая мультифокальная лейкоэнцефалопатия после ритуксимаба у пациента с рецидивом фолликулярной лимфомы и низким уровнем IgG и низким количеством CD4+ лимфоцитов. Лейк-лимфома. 2008;49: 2367–2369. [PubMed] [Google Scholar]
32. Cinque P, Koralnik IJ, Clifford DB. Развивающееся лицо прогрессирующей многоочаговой лейкоэнцефалопатии, связанной с вирусом иммунодефицита человека: определение согласованной терминологии.![]() J Нейровирол. 2003; 9 (прил. 1): 88–92. [PubMed] [Google Scholar]
J Нейровирол. 2003; 9 (прил. 1): 88–92. [PubMed] [Google Scholar]
33. Weber F, Goldmann C, Kramer M, et al. Клеточный и гуморальный иммунный ответ при прогрессирующей мультифокальной лейкоэнцефалопатии. Энн Нейрол. 2001; 49: 636–642. [PubMed] [Google Scholar]
34. Houff SA, Major EO, Katz DA, et al. Участие зараженных вирусом JC мононуклеарных клеток костного мозга и селезенки в патогенезе прогрессирующей мультифокальной лейкоэнцефалопатии. N Engl J Med. 1988;318:301–305. [PubMed] [Google Scholar]
35. Stasi R, Del Poeta G, Stipa E, et al. Ответ на терапию, направленную на истощение В-клеток ритуксимабом, устраняет аномалии субпопуляций Т-клеток у пациентов с идиопатической тромбоцитопенической пурпурой. Кровь. 2007;110:2924–2930. [PubMed] [Google Scholar]
36. Нарула С., ЛаРоса Д.Ф., Камун М., Далмау Дж., Левинсон А.И. Прогрессирующая мультифокальная лейкоэнцефалопатия у пациента с общим вариабельным иммунодефицитом и аномальным распределением субпопуляции CD8+ Т-клеток. Энн Аллергия Астма Иммунол. 2007;98: 483–489. [PubMed] [Google Scholar]
Энн Аллергия Астма Иммунол. 2007;98: 483–489. [PubMed] [Google Scholar]
37. Терамото Т., Канеко Х., Фунато М. и соавт. Прогрессирующая мультифокальная лейкоэнцефалопатия у больного с Х-сцепленной агаммаглобулинемией. Scand J Infect Dis. 2003; 35: 909–910. [PubMed] [Google Scholar]
38. Sabath BF, Major EO. Трафик вируса JC из очагов первичной инфекции в головной мозг: путь к прогрессирующей мультифокальной лейкоэнцефалопатии. J заразить дис. 2002; 186 ([приложение 2]): S180–S186. [PubMed] [Google Scholar]
39. Bonig H, Wundes A, Chang KH, Lucas S, Papayannopoulou T. Повышенное количество циркулирующих гемопоэтических стволовых клеток/клеток-предшественников хронически поддерживается у пациентов, получающих CD49.d блокирующее антитело натализумаб. Кровь. 2008; 111:3439–3441. [Бесплатная статья PMC] [PubMed] [Google Scholar]
40. Линдберг Р.Л., Ахтнихтс Л., Хоффманн Ф., Куле Дж., Каппос Л. Натализумаб изменяет профили экспрессии транскрипции субпопуляций клеток крови у пациентов с рассеянным склерозом. J Нейроиммунол. 2008; 194: 153–164. [PubMed] [Google Scholar]
J Нейроиммунол. 2008; 194: 153–164. [PubMed] [Google Scholar]
41. Зорен Ф., Тутзарис Д., Кларнер В., Хартунг Х.П., Кизиер Б., Хаас Р. Моноклональное антитело против VLA-4 натализумаб мобилизует гемопоэтические клетки-предшественники CD34+ у людей. Кровь. 2008;111:3893–3895. [PubMed] [Google Scholar]
42. Gibson PE, Gardner SD, Field AM. Использование молекулярного зонда для обнаружения ДНК JCV непосредственно в материале головного мозга человека. J Med Virol. 1986; 18:87–95. [PubMed] [Google Scholar]
43. Fong IW, Britton CB, Luinstra KE, Toma E, Mahony JB. Диагностическое значение обнаружения ДНК вируса JC в спинномозговой жидкости больных прогрессирующей мультифокальной лейкоэнцефалопатией. Дж. Клин Микробиол. 1995; 33: 484–486. [Бесплатная статья PMC] [PubMed] [Google Scholar]
44. Dubois V, Moret H, Lafon ME, et al. Распространенность виремии вируса JC у ВИЧ-инфицированных пациентов с неврологическими расстройствами или без них: проспективное исследование. J Нейровирол. 1998; 4: 539–544. [PubMed] [Google Scholar]
J Нейровирол. 1998; 4: 539–544. [PubMed] [Google Scholar]
45. Delbue S, Guerini FR, Mancuso R, et al. Виремия вируса JC у пациентов с итальянским рассеянным склерозом, получавших и не получавших интерферон-бета, и у здоровых лиц контрольной группы. J Нейровирол. 2007; 13:73–77. [PubMed] [Google Scholar]
46. Engsig FN, Hansen AB, Omland LH, et al. Заболеваемость, клиническая картина и исход прогрессирующей мультифокальной лейкоэнцефалопатии у ВИЧ-инфицированных пациентов в эпоху высокоактивной антиретровирусной терапии: общенациональное когортное исследование. J заразить дис. 2009 г.;199:77–83. [PubMed] [Google Scholar]
47. Бергер Дж. Р., майор Е.О. Прогрессирующая мультифокальная лейкоэнцефалопатия. Семин Нейрол. 1999; 19: 193–200. [PubMed] [Google Scholar]
48. Laszlo D, Bassi S, Andreola G, et al. Субпопуляции периферических Т-лимфоцитов у пациентов, получавших комбинированную терапию ритуксимабом и хлорамбуцилом при индолентной НХЛ. Энн Хематол. 2006; 85: 813–814. [PubMed] [Google Scholar]
[PubMed] [Google Scholar]
49. Брукс Б.Р., Уокер Д.Л. Прогрессирующая мультифокальная лейкоэнцефалопатия. Нейрол клин. 1984;2:299–313. [PubMed] [Google Scholar]
50. Garcia-Suarez J, de Miguel D, Krsnik I, Banas H, Arribas I, Burgaleta C. Изменения в естественном течении прогрессирующей многоочаговой лейкоэнцефалопатии при ВИЧ-отрицательных лимфопролиферативных заболеваниях: влияние новые методы лечения. Am J Гематол. 2005; 80: 271–281. [PubMed] [Google Scholar]
51. Winstein K. Biogen ссылается на новую неудачу для лекарств от рассеянного склероза. Уолл Стрит Джорнал. 2008, 16 декабря;: D2. [Google Scholar]
52. Kappos L, Bates D, Hartung HP, et al. Лечение рассеянного склероза натализумабом: рекомендации по отбору пациентов и мониторингу. Ланцет Нейрол. 2007; 6: 431–441. [PubMed] [Академия Google]
53. Wysowski D, Bozic C. Gaithersburg, MD: 2006. План действий по минимизации рисков и план управления рисками при применении натализумаба: заседание Консультативного комитета по наркотикам Управления по санитарному надзору за качеством пищевых продуктов и медикаментов. [Google Scholar]
[Google Scholar]
54. FDA. Предупреждение FDA: информация для медицинских работников: инъекция натализумаба для внутривенного введения. [По состоянию на 2 марта 2009 г.]; август 2008 г.; http://www.fda.gov/cder/drug/InfoSheets/HCP/natalizumab-2008HCP.htm.
55. Дженентех. Руководство по лекарствам Ритуксан (ритуксимаб) [По состоянию на 2 марта 2009 г.]; 2008 г. http://www.gene.com/gene/products/information/pdf/rituxan_med_guide.pdf.
56. Coiffier B, Lepage E, Briere J, et al. Химиотерапия CHOP плюс ритуксимаб по сравнению с монотерапией CHOP у пожилых пациентов с диффузной крупноклеточной В-клеточной лимфомой. N Engl J Med. 2002; 346: 235–242. [PubMed] [Google Scholar]
57. Feugier P, Van Hoof A, Sebban C, et al. Отдаленные результаты исследования R-CHOP в лечении пожилых пациентов с диффузной крупноклеточной В-клеточной лимфомой: исследование Groupe d’Etude des Lymphomes de l’Adulte. Дж. Клин Онкол. 2005; 23:4117–4126. [PubMed] [Академия Google]
58. Маркус Р., Имри К., Солал-Селиньи П. и соавт. Исследование III фазы R-CVP по сравнению с монотерапией циклофосфамидом, винкристином и преднизоном у пациентов с ранее не леченной распространенной фолликулярной лимфомой. Дж. Клин Онкол. 2008; 26: 4579–4586. [PubMed] [Google Scholar]
Маркус Р., Имри К., Солал-Селиньи П. и соавт. Исследование III фазы R-CVP по сравнению с монотерапией циклофосфамидом, винкристином и преднизоном у пациентов с ранее не леченной распространенной фолликулярной лимфомой. Дж. Клин Онкол. 2008; 26: 4579–4586. [PubMed] [Google Scholar]
59. Pfreundschuh M, Trumper L, Osterborg A, et al. CHOP-подобная химиотерапия плюс ритуксимаб по сравнению с CHOP-подобной химиотерапией только у молодых пациентов с диффузной крупноклеточной В-крупноклеточной лимфомой с хорошим прогнозом: рандомизированное контролируемое исследование, проведенное Международной исследовательской группой MabThera (MInT). Ланцет Онкол. 2006;7:379–391. [PubMed] [Google Scholar]
60. Fu K, Weisenburger DD, Choi WW, et al. Добавление ритуксимаба к стандартной химиотерапии улучшает выживаемость подтипов диффузной В-крупноклеточной лимфомы как с зародышевым центром В-клеток, так и с негерминальным центром В-клеток. Дж. Клин Онкол. 2008; 26: 4587–4594. [PubMed] [Google Scholar]
61. Arnold DM, Dentali F, Crowther MA, et al. Систематический обзор: эффективность и безопасность ритуксимаба у взрослых с идиопатической тромбоцитопенической пурпурой. Энн Интерн Мед. 2007; 146: 25–33. [PubMed] [Академия Google]
Arnold DM, Dentali F, Crowther MA, et al. Систематический обзор: эффективность и безопасность ритуксимаба у взрослых с идиопатической тромбоцитопенической пурпурой. Энн Интерн Мед. 2007; 146: 25–33. [PubMed] [Академия Google]
62. Cohen SB, Emery P, Greenwald MW, et al. Ритуксимаб при ревматоидном артрите, рефрактерном к терапии факторами некроза опухоли: результаты многоцентрового рандомизированного двойного слепого плацебо-контролируемого исследования III фазы по оценке первичной эффективности и безопасности через 24 недели. Ревмирующий артрит. 2006; 54: 2793–2806. [PubMed] [Google Scholar]
63. Hauser SL, Waubant E, Arnold DL, et al. Истощение В-клеток с помощью ритуксимаба при рецидивирующем-ремиттирующем рассеянном склерозе. N Engl J Med. 2008; 358: 676–688. [PubMed] [Академия Google]
64. Ng KP, Cambridge G, Leandro MJ, Edwards JC, Ehrenstein M, Isenberg DA. Терапия истощения В-клеток при системной красной волчанке: долгосрочное наблюдение и предикторы ответа. Энн Реум Дис. 2007; 66: 1259–1262. [Бесплатная статья PMC] [PubMed] [Google Scholar]
Энн Реум Дис. 2007; 66: 1259–1262. [Бесплатная статья PMC] [PubMed] [Google Scholar]
65. Bennett CL, Nebeker JR, Lyons EA, et al. Проект «Исследования нежелательных явлений и отчетов о лекарственных препаратах» (RADAR). ДЖАМА. 2005; 293:2131–2140. [PubMed] [Google Scholar]
66. Bennett CL, Nebeker JR, Yarnold PR, et al. Оценка серьезных побочных реакций на лекарственные препараты: программа упреждающего фармаконадзора (RADAR) в сравнении с мероприятиями по обеспечению безопасности, проводимыми Управлением по санитарному надзору за качеством пищевых продуктов и медикаментов и производителями фармацевтических препаратов. Arch Intern Med. 2007; 167:1041–1049.. [PubMed] [Google Scholar]
67. Holman RC, Torok TJ, Belay ED, Janssen RS, Schonberger LB. Прогрессирующая многоочаговая лейкоэнцефалопатия в США, 1979-1994 гг.: повышенная смертность, связанная с ВИЧ-инфекцией. Нейроэпидемиология. 1998; 17: 303–309. [PubMed] [Google Scholar]
68. Кавано А., Маттесон Э. Горячая линия: Ритуксимаб и прогрессирующая многоочаговая лейкоэнцефалопатия. [По состоянию на 2 марта 2009 г.] Американский колледж ревматологии. 2008 г. http://www.rheumatology.org/publications/hotline/0107leuko.asp.
Горячая линия: Ритуксимаб и прогрессирующая многоочаговая лейкоэнцефалопатия. [По состоянию на 2 марта 2009 г.] Американский колледж ревматологии. 2008 г. http://www.rheumatology.org/publications/hotline/0107leuko.asp.
Хромосома 17: MedlinePlus Genetics
URL этой страницы: https://medlineplus.gov/genetics/chromosome/17/
Чтобы использовать функции обмена на этой странице, включите JavaScript.
Описание
В норме у человека в каждой клетке 46 хромосом, разделенных на 23 пары. Две копии хромосомы 17, по одной копии, унаследованной от каждого родителя, образуют одну из пар. Хромосома 17 охватывает около 83 миллионов строительных блоков ДНК (пар оснований) и составляет от 2,5 до 3 процентов всей ДНК в клетках.
Идентификация генов на каждой хромосоме является активной областью генетических исследований. Поскольку исследователи используют разные подходы для прогнозирования количества генов на каждой хромосоме, предполагаемое количество генов варьируется.
Заболевания, связанные с хромосомными изменениями
Следующие хромосомные заболевания связаны с изменениями в структуре или числе копий хромосомы 17.
Синдром делеции 17q12
Синдром делеции 17q12 — это состояние, возникающее в результате делеции небольшого фрагмента хромосомы 17 в каждой клетке. Признаки и симптомы синдрома делеции 17q12 могут включать аномалии почек и мочевыделительной системы, форму диабета, называемую диабетом зрелого возраста 5-го типа (MODY5), задержку развития, умственную отсталость и поведенческие или психические расстройства. У некоторых женщин с этим хромосомным изменением наблюдается синдром Майера-Рокитанского-Кюстера-Хаузера, который характеризуется недоразвитием или отсутствием влагалища и матки. Особенности, связанные с синдромом делеции 17q12, сильно различаются даже среди пораженных членов одной семьи.
У большинства людей с синдромом делеции 17q12 отсутствует около 1,4 миллиона строительных блоков ДНК (пар оснований), также обозначаемых как 1,4 мегабазы (Мб), на длинном (q) плече хромосомы в позиции, обозначенной q12. Это тот же участок хромосомы 17, который аномально копируется (дублируется) у людей с дупликацией 17q12 (описано ниже). Этот сегмент хромосомы окружен короткими повторяющимися последовательностями ДНК, которые делают его склонным к перестройке во время клеточного деления. Перестройка может привести к недостающим или дополнительным копиям ДНК в 17q12.
Сегмент, который чаще всего делетируется у людей с синдромом делеции 17q12, включает 15 генов. Некоторые из особенностей, связанных с этим состоянием, вероятно, являются результатом потери двух из этих генов, HNF1B и LHX1
 Потеря других генов в удаленной области также может влиять на признаки и симптомы синдрома делеции 17q12.
Потеря других генов в удаленной области также может влиять на признаки и симптомы синдрома делеции 17q12.Подробнее об этом заболевании
Дупликация 17q12
Дупликация 17q12 представляет собой хромосомное изменение, при котором небольшой участок хромосомы 17 аномально копируется в каждой клетке. Признаки и симптомы, связанные с этим удвоением, значительно различаются даже среди членов одной семьи. У некоторых людей с дупликацией нет явных признаков или симптомов, или они очень слабо выражены. Другие люди могут иметь умственную отсталость, задержку развития и широкий спектр физических аномалий.
У большинства людей с дупликацией 17q12 имеется дополнительная копия ДНК размером около 1,4 Мб в положении q12 на хромосоме 17. Это тот же участок хромосомы 17, который удален у людей с синдромом делеции 17q12 (описанным выше). Этот сегмент хромосомы склонен к перестройке во время клеточного деления, что может привести к дополнительным или отсутствующим копиям ДНК в 17q12.
Дублированный сегмент 17q12 включает не менее 15 генов. Неясно, какие из этих генов, если они присутствуют более чем в одной копии, способствуют умственной отсталости, задержке развития и другим признакам, которые могут быть связаны с дупликацией 17q12. Поскольку у некоторых людей с этой дупликацией нет очевидных интеллектуальных или физических проблем, исследователи подозревают, что дополнительные генетические факторы могут влиять на наличие у человека признаков и симптомов, связанных с хромосомными изменениями.
Подробнее об этом заболевании
Острый промиелоцитарный лейкоз
Тип рака крови, известный как острый промиелоцитарный лейкоз, вызывается перестройкой (транслокацией) генетического материала между хромосомами 15 и 17. Эта транслокация, обозначаемая как t(15;17), сливает часть Ген PML из хромосомы 15 с частью гена RARA из хромосомы 17. Эта мутация приобретается в течение жизни человека и присутствует только в определенных клетках. Этот тип генетического изменения, называемый соматической мутацией, не передается по наследству. Транслокацию t(15;17) называют сбалансированной реципрокной транслокацией, поскольку фрагменты хромосом обмениваются друг с другом (реципрокная) и генетический материал не приобретается и не теряется (сбалансированная). Белок, полученный из этого слитого гена, известен как PML-RARα.
Этот тип генетического изменения, называемый соматической мутацией, не передается по наследству. Транслокацию t(15;17) называют сбалансированной реципрокной транслокацией, поскольку фрагменты хромосом обмениваются друг с другом (реципрокная) и генетический материал не приобретается и не теряется (сбалансированная). Белок, полученный из этого слитого гена, известен как PML-RARα.
Белок PML-RARα функционирует иначе, чем белковые продукты нормальных генов PML и RARA . Ген RARA на хромосоме 17 предоставляет инструкции для создания фактора транскрипции, называемого альфа-рецептором ретиноевой кислоты (RARα). Фактор транскрипции — это белок, который прикрепляется (связывается) к определенным областям ДНК и помогает контролировать активность (транскрипцию) определенных генов. В норме белок RARα контролирует активность генов, важных для созревания (дифференциации) незрелых лейкоцитов за пределами определенной стадии, называемой промиелоцитом. Ген PML на хромосоме 15 предоставляет инструкции для белка, который действует как супрессор опухоли, что означает, что он предотвращает слишком быстрый или неконтролируемый рост и деление клеток.
Подробнее об этом заболевании
Болезнь Шарко-Мари-Тута
Дупликация небольшого фрагмента хромосомы 17 в положении p12, который включает ген PMP22 , вызывает большинство случаев заболевания, называемого болезнью Шарко-Мари-Тута. Когда это заболевание вызвано изменениями, затрагивающими ген PMP22 , оно называется болезнью Шарко-Мари-Тута типа 1А или CMT1A. Болезнь Шарко-Мари-Тута повреждает периферические нервы, соединяющие головной и спинной мозг с мышцами и сенсорными клетками, которые обнаруживают такие ощущения, как прикосновение, боль, тепло и звук.
Белок, полученный из гена PMP22 , является компонентом миелина, защитного вещества, покрывающего нервы и способствующего эффективной передаче нервных импульсов. Прежде чем они станут частью миелина, новообразованные белки PMP22 обрабатываются и упаковываются в специализированные клеточные структуры, называемые эндоплазматическим ретикулумом и аппаратом Гольджи. Завершение этих этапов обработки и упаковки имеет решающее значение для правильного развития, поддержания и функционирования миелина.
Дополнительная копия гена PMP22 , возникающая в результате дупликации, приводит к перепроизводству белка PMP22. Исследования показывают, что избыток белка PMP22 может подавлять способность клеток правильно обрабатывать его, что приводит к накоплению необработанного, нефункционального белка. Это накопление может нарушить образование миелина и привести к нестабильности и потере миелина (демиелинизации).
Подробнее об этом заболевании
Выступающая дерматофибросаркома
Транслокация генетического материала между хромосомами 17 и 22, обозначаемая как t(17;22), вызывает редкий тип рака кожи, известный как выбухающая дерматофибросаркома. Эта транслокация объединяет часть гена COL1A1 из хромосомы 17 с частью гена PDGFB из хромосомы 22. Транслокация обнаруживается на одной или нескольких дополнительных хромосомах, которые могут быть линейными или кольцевыми. Когда они кольцевые, дополнительные хромосомы известны как нештатные кольцевые хромосомы. Эта мутация приобретается в течение жизни человека и присутствует только в определенных клетках. Этот тип генетического изменения, называемый соматической мутацией, не передается по наследству.
Слитый ген COL1A1-PDGFB содержит инструкции по созданию комбинированного (слитого) белка, который, по мнению исследователей, в конечном счете функционирует подобно активному белку PDGFB. При транслокации ген PDGFB теряет часть своей ДНК, которая ограничивает его активность, и продукция слитого белка COL1A1-PDGFB контролируется последовательностями гена COL1A1 . В результате слияние генов приводит к продукции большего количества активного белка PDGFB, чем обычно. Активный белок PDGFB сигнализирует о росте и делении клеток (пролиферации) и созревании (дифференцировке). Избыток белка PDGFB аномально стимулирует пролиферацию и дифференцировку клеток, что приводит к образованию опухоли, наблюдаемой при выбухающей дерматофибросаркоме.
Подробнее об этом заболевании
Синдром Кулена-де Фриза
Делеция небольшого количества генетического материала (микроделеция) на хромосоме 17 может вызвать синдром Кулена-де Фриза. Это расстройство характеризуется задержкой развития, умственной отсталостью, веселым и общительным нравом и разнообразными физическими отклонениями.
Это расстройство характеризуется задержкой развития, умственной отсталостью, веселым и общительным нравом и разнообразными физическими отклонениями.
У большинства людей с синдромом Кулена-де Фриза отсутствует последовательность примерно из 500 000 пар оснований, также обозначаемая как 500 килобаз (т.п.н.), в положении q21.31 на хромосоме 17. Точный размер делеции варьируется у разных людей, но он содержит не менее шести генов, в том числе КАНСЛ1 . Эта делеция затрагивает одну из двух копий хромосомы 17 в каждой клетке.
Поскольку мутации в гене KANSL1 вызывают те же признаки и симптомы, что и делеция, исследователи пришли к выводу, что потеря этого гена объясняет особенности синдрома Кулена-де Фриза. Белок, полученный из гена KANSL1 , участвует в контроле активности других генов и играет важную роль в развитии и функционировании многих частей тела. Хотя потеря этого гена нарушает нормальное развитие и функцию, его связь со специфическими чертами синдрома Кулена-де Фриза неясна.
Хотя синдром Кулена-де Фриза обычно не передается по наследству, у большинства людей с заболеванием, вызванным делецией, по крайней мере один из родителей имел распространенный вариант области q21.31 хромосомы 17, называемый линией h3. Этот вариант встречается у 20 процентов людей европейского и ближневосточного происхождения, хотя в других популяциях он встречается редко. В линии h3 сегмент ДНК длиной 900 т.п.н., который включает область, делетированную в большинстве случаев синдрома Кулена-де Фриза, подвергся инверсии. Инверсия включает два разрыва в хромосоме; полученный фрагмент ДНК переворачивается и повторно вставляется в хромосому.
Люди с родословной h3 не имеют проблем со здоровьем, связанных с инверсией. Однако генетический материал может быть потерян или продублирован, когда инверсия передается следующему поколению. Исследователи полагают, что родительская инверсия, вероятно, необходима для того, чтобы у ребенка была микроделеция 17q21.31, наиболее часто связанная с синдромом Кулена-де Фриза, но также считается, что другие неизвестные факторы также играют роль. Таким образом, несмотря на то, что инверсия очень распространена, только у очень небольшого процента родителей с инверсией есть ребенок, страдающий синдромом Кулена-де Фриза.
Таким образом, несмотря на то, что инверсия очень распространена, только у очень небольшого процента родителей с инверсией есть ребенок, страдающий синдромом Кулена-де Фриза.
Подробнее об этом заболевании
Синдром Миллера-Дикера
Синдром Миллера-Дикера вызывается делецией генетического материала вблизи конца короткого (p) плеча хромосомы 17. Признаки и симптомы синдрома Миллера-Дикера связаны с потерей нескольких генов в этой области. Размер делеции варьируется среди пострадавших людей. Потеря определенного гена на 17-й хромосоме, называемого PAFAh2B1 , отвечает за характерный признак синдрома — лиссэнцефалию, проблему развития мозга, при которой поверхность мозга аномально гладкая. Эта аномалия головного мозга вызывает тяжелую умственную отсталость, задержку развития, судороги, аномальную ригидность мышц (спасичность), слабый мышечный тонус (гипотонию) и трудности с кормлением. Потеря другого гена, называемого YWHAE в той же области хромосомы 17 увеличивает тяжесть лиссэнцефалии у людей с синдромом Миллера-Дикера. Дополнительные гены в удаленной области вносят свой вклад в различные черты этого расстройства.
Дополнительные гены в удаленной области вносят свой вклад в различные черты этого расстройства.
Подробнее об этом заболевании
Синдром Потоцкого-Лупски
Синдром Потоцкого-Лупски возникает в результате дупликации небольшого фрагмента хромосомы 17 в каждой клетке, особенно в области короткого (p) плеча, обозначенной p11.2. Это состояние характеризуется задержкой развития, умственной отсталостью от легкой до умеренной, поведенческими проблемами, включая расстройство аутистического спектра (которое влияет на социальное взаимодействие и общение), нарушениями сна и другими проблемами со здоровьем.
Примерно у двух третей больных размер дублированного сегмента составляет приблизительно 3,7 Мб. (Отсутствие копии этого сегмента вызывает синдром Смита-Магениса, описанный ниже.) В оставшейся трети случаев дупликация больше или меньше, от менее 1 Мб до почти 20 Мб. Все эти дупликации затрагивают одну из двух копий хромосомы 17 в каждой клетке.
Хотя дуплицированная область содержит несколько генов, исследователи полагают, что наличие дополнительной копии одного конкретного гена RAI1 лежит в основе многих характерных признаков синдрома Потоцкого-Лупски. Все известные дупликации, вызывающие это состояние, содержат этот ген. Ген RAI1 предоставляет инструкции по созданию белка, который помогает регулировать активность (экспрессию) других генов. Хотя большинство генов, регулируемых белком RAI1, не были идентифицированы, этот белок, по-видимому, контролирует экспрессию нескольких генов, участвующих в дневных (циркадных) ритмах, таких как цикл сон-бодрствование. Белок RAI1, по-видимому, также играет роль в развитии головного мозга и костей головы и лица. Исследования показывают, что дублирование увеличивает количество белка RAI1, что нарушает экспрессию генов, влияющих на циркадные ритмы. Эти изменения могут быть причиной нарушений сна, возникающих при синдроме Потоцкого-Лупски. Слишком много белка RAI1 может также нарушить развитие мозга, что может объяснить задержку развития, умственную отсталость, поведенческие проблемы и другие неврологические особенности этого состояния. Также может быть затронуто развитие костей головы и лица, что приводит к тонким лицевым различиям у людей с синдромом Потоцкого-Лупски.
Также может быть затронуто развитие костей головы и лица, что приводит к тонким лицевым различиям у людей с синдромом Потоцкого-Лупски.
Подробнее об этом заболевании
Синдром Смита-Магениса
Синдром Смита-Магениса обычно возникает в результате делеции небольшого фрагмента хромосомы 17 в каждой клетке, особенно в области короткого (p) плеча, обозначенной p11.2. Это нарушение развития затрагивает многие части тела. Основные признаки этого состояния включают умственную отсталость от легкой до умеренной степени, задержку речевых и языковых навыков, характерные черты лица, нарушения сна и поведенческие проблемы.
Чаще всего сегмент хромосомы, удаленный при синдроме Смита-Магениса, совпадает с сегментом хромосомы, который дублируется при синдроме Потоцкого-Лупски (описан выше). Иногда делеция больше или меньше. Все делеции затрагивают одну из двух копий хромосомы 17 в каждой клетке.
Исследователи полагают, что потеря функции гена RAI1 объясняет многие признаки и симптомы синдрома Смита-Магениса. Все известные делеции, вызывающие это состояние, содержат этот ген. Исследования показывают, что делеция приводит к уменьшению количества белка RAI1 в клетках, что нарушает экспрессию генов, участвующих в циркадных ритмах. Эти изменения могут быть причиной нарушений сна, возникающих при синдроме Смита-Магениса. Непонятно, как потеря одного экземпляра 9Ген 0161 RAI1 приводит к другим физическим, психическим и поведенческим проблемам, связанным с этим заболеванием. Вполне вероятно, что потеря других генов в удаленной области также влияет на признаки и симптомы; роль этих генов изучается.
Все известные делеции, вызывающие это состояние, содержат этот ген. Исследования показывают, что делеция приводит к уменьшению количества белка RAI1 в клетках, что нарушает экспрессию генов, участвующих в циркадных ритмах. Эти изменения могут быть причиной нарушений сна, возникающих при синдроме Смита-Магениса. Непонятно, как потеря одного экземпляра 9Ген 0161 RAI1 приводит к другим физическим, психическим и поведенческим проблемам, связанным с этим заболеванием. Вполне вероятно, что потеря других генов в удаленной области также влияет на признаки и симптомы; роль этих генов изучается.
Подробнее об этом заболевании
Синдром Юань-Харела-Лупски
Удвоение небольшого фрагмента хромосомы 17 в области, обозначенной p12-11.2, может вызвать синдром Юань-Харела-Лупски (ЮХАЛ), который характеризуется множественными неврологическими проблемами, сходными с таковыми при Потоцком-Лупски. синдром (описан выше) и CMT1A (описан выше). При синдроме ЮХАЛ дуплицированный сегмент имеет размер от 3 до 20 Мб и всегда содержит 9 нуклеотидов. 0161 RAI1 и гены PMP22 ; он также может включать дополнительные гены. Некоторые особенности синдрома ЮХАЛ, такие как задержка развития и поведенческие проблемы, вероятно, вызваны дополнительной копией гена RAI1 . Другие признаки, в том числе мышечная слабость и снижение чувствительности к прикосновению, теплу и холоду в голенях и ступнях, вероятно, связаны с дупликацией гена PMP22 .
0161 RAI1 и гены PMP22 ; он также может включать дополнительные гены. Некоторые особенности синдрома ЮХАЛ, такие как задержка развития и поведенческие проблемы, вероятно, вызваны дополнительной копией гена RAI1 . Другие признаки, в том числе мышечная слабость и снижение чувствительности к прикосновению, теплу и холоду в голенях и ступнях, вероятно, связаны с дупликацией гена PMP22 .
Подробнее об этом заболевании
Другие хромосомные заболевания
Другие изменения числа или структуры хромосомы 17 могут иметь различные последствия, включая умственную отсталость, задержку развития, характерные черты лица, слабый мышечный тонус (гипотонию) и низкий рост. Эти изменения включают дополнительный участок 17-й хромосомы в каждой клетке (частичная трисомия 17), недостающий сегмент хромосомы в каждой клетке (частичная моносомия 17) и кольцевую структуру, называемую кольцевой хромосомой 17. Кольцевые хромосомы возникают при разрыве хромосомы. в двух местах, а концы хромосомных плеч сливаются вместе, образуя кольцевую структуру.
в двух местах, а концы хромосомных плеч сливаются вместе, образуя кольцевую структуру.
Другие виды рака
Изменения в хромосоме 17 были выявлены при нескольких дополнительных типах рака. Эти генетические изменения являются соматическими, то есть приобретаются в течение жизни человека и присутствуют только в определенных клетках. Особая хромосомная аномалия, называемая изохромосомой 17q, часто встречается при некоторых видах рака. Эта аномальная версия хромосомы 17 имеет два длинных (q) плеча вместо одного длинного и одного короткого (p) плеча. В результате в хромосоме есть лишняя копия одних генов и отсутствуют копии других генов.
Изохромосома 17q обычно обнаруживается при раке кроветворной ткани, называемом хроническим миелоидным лейкозом (ХМЛ). Он также был обнаружен в некоторых солидных опухолях, в том числе в опухоли головного мозга, называемой медуллобластомой, и в опухолях головного и спинного мозга, известных как примитивные нейроэктодермальные опухоли. Хотя изохромосома 17q, вероятно, играет роль как в развитии, так и в прогрессировании этих видов рака, конкретные генетические изменения, связанные с ростом рака, неизвестны.
Хотя изохромосома 17q, вероятно, играет роль как в развитии, так и в прогрессировании этих видов рака, конкретные генетические изменения, связанные с ростом рака, неизвестны.
Дополнительная информация и ресурсы
Дополнительные ресурсы NIH
- Национальный институт исследования генома человека: Хромосомные аномалии
Научные статьи в PubMed
- PubMed
Ссылки
- Барбути А., Станкевич П., Нусбаум С., Куомо С., Кук А., Хёглунд М., Йоханссон Б., Хагемейер А., Парк С.С., Мительман Ф., Лупски Дж.Р., Фиоретос Т. Область точки останова наиболее распространенной изохромосомы i(17q) при неоплазии человека характеризуется сложная геномная архитектура с большими палиндромными малокопийными повторами. Ам Дж Хам Жене. 2004 г., январь; 74 (1): 1–10. Epub 2003, 8 декабря. Цитирование в PubMed или бесплатная статья в PubMed Central
- Кардозо К., Левентер Р.Дж., Уорд Х.Л.
 , Тойо-Ока К., Чанг Дж., Гросс А., Мартин К.Л.,
Аллансон Дж., Пильц Д.Т., Олни А.Х., Мутчиник О.М., Хироцунэ С., Уиншоу-Борис А.,
Добинс В.Б., Ледбеттер Д.Х. Уточнение критической области размером 400 т.п.н. позволяет определить генотип
дифференциация изолированной лиссэнцефалии, синдрома Миллера-Дикера и др.
фенотипы вторичны по отношению к делеции 17p13.3. Am J Hum Genet. 2003 г.
Апр; 72 (4): 918-30. Epub 2003 Mar 5. Цитирование в PubMed или бесплатная статья в PubMed Central
, Тойо-Ока К., Чанг Дж., Гросс А., Мартин К.Л.,
Аллансон Дж., Пильц Д.Т., Олни А.Х., Мутчиник О.М., Хироцунэ С., Уиншоу-Борис А.,
Добинс В.Б., Ледбеттер Д.Х. Уточнение критической области размером 400 т.п.н. позволяет определить генотип
дифференциация изолированной лиссэнцефалии, синдрома Миллера-Дикера и др.
фенотипы вторичны по отношению к делеции 17p13.3. Am J Hum Genet. 2003 г.
Апр; 72 (4): 918-30. Epub 2003 Mar 5. Цитирование в PubMed или бесплатная статья в PubMed Central - Чен К.С., Маниан П., Койт Т., Потоцкий Л., Чжао К., Чиноль А.С., Ли К.С., Лупски младший Гомологическая рекомбинация кластера генов фланкирующих повторов является механизмом распространенный синдром делеции смежных генов. Нат Жене. 1997 окт; 17(2):154-63. Цитата в PubMed
- de Thé H, Lavau C, Marchio A, Chomienne C, Degos L, Dejean A. PML-RAR
мРНК альфа-слияния, генерируемая транслокацией t(15;17) при остром промиелоцитарном
лейкемия кодирует функционально измененный RAR.
 Клетка. 1991 23 августа; 66(4):675-84.
Цитата в PubMed
Клетка. 1991 23 августа; 66(4):675-84.
Цитата в PubMed - Дюпон С., Пипирас Э., Шанто-Бастаро С., Верлоэс А., Бауманн С., Вольф Дж. П., Benzacken B. CGH и прямая диагностика мозаичной структурной хромосомной аномалии: описание мозаичной кольцевой хромосомы 17 и обзор литература. Eur J Hum Genet. 2003 г., июнь; 11 (6): 452-6. Цитата в PubMed
- Элиас Р.С., Галера М.Ф., Шнабель Б., Брионес М.Р., Борри М.Л., Липай М., Карвальейра Г., Брунони Д., Мелараньо М.И. Делеция 17p13 и мутация гена LIS1 у изолированных последовательность лиссэнцефалии. Педиатр Нейрол. 2006 г., июль; 35 (1): 42-6. Цитата на PubMed
- Ensembl Human Map View
- Гилберт Ф. Гены болезней и хромосомы: карты болезней генома человека. Хромосома 17. Генетический тест. 1998;2(4):357-81. Цитата в PubMed
- Греко А., Фузетти Л., Вилла Р., Соцци Г., Минолетти Ф., Маури П., Пьеротти М.А.
Трансформирующая активность химерной последовательности, образованной слиянием коллагена
ген COL1A1 и ген b-цепи тромбоцитарного фактора роста в
выбухающая дерматофибросаркома.
 Онкоген. 1998 10 сентября; 17 (10): 1313-9. Цитата в PubMed
Онкоген. 1998 10 сентября; 17 (10): 1313-9. Цитата в PubMed - Katona I, Wu X, Feely SM, Sottile S, Siskind CE, Miller LJ, Shy ME, Li J. Экспрессия PMP22 в миелине кожных нервов у пациентов с CMT1A. Мозг. 2009 г. Июль; 132 (Pt 7): 1734-40. doi: 10.1093/мозг/awp113. Epub 2009, 15 мая. Цитирование в PubMed или бесплатная статья на PubMed Central
- Кулен Д.А., Крамер Дж.М., Невелинг К., Ниллесен В.М., Мур-Бартон Х.Л., Элмсли Ф.В., Тутэн А., Амиэль Дж., Малан В., Цай А.С., Чунг С.В., Гилиссен С., Вервиль Э.Т., Мартенс S, Feuth T, Bongers EM, de Vries P, Scheffer H, Vissers LE, de Brouwer AP, Бруннер Х.Г., Вельтман Дж.А., Шенк А., Интема Х.Г., де Врис Б.Б. Мутации в Ген-модификатор хроматина KANSL1 вызывает синдром микроделеции 17q21.31. Нат Жене. 2012 29 апр.;44(6):639-41. doi: 10.1038/ng.2262. Цитата в PubMed
- Кулен Д.А., Виссерс Л.Е., Пфундт Р., де Леу Н., Найт С.Дж., Риган Р., Кой Р.Ф.,
Рейньерс Э., Романо С.
 , Фичера М., Шинцель А., Баумер А., Андерлид Б.М., Шуманс Дж.,
Кноерс Н.В., ван Кессель А.Г., Систерманс Э.А., Вельтман Дж.А., Бруннер Х.Г., де Врис Б.Б. А
новый синдром микроделеции хромосомы 17q21.31, связанный с распространенной инверсией
полиморфизм. Нат Жене. 2006 г., сен; 38 (9): 999-1001. Epub 2006, 13 августа. Цитирование на PubMed
, Фичера М., Шинцель А., Баумер А., Андерлид Б.М., Шуманс Дж.,
Кноерс Н.В., ван Кессель А.Г., Систерманс Э.А., Вельтман Дж.А., Бруннер Х.Г., де Врис Б.Б. А
новый синдром микроделеции хромосомы 17q21.31, связанный с распространенной инверсией
полиморфизм. Нат Жене. 2006 г., сен; 38 (9): 999-1001. Epub 2006, 13 августа. Цитирование на PubMed - Меффорд Х.К., Клауин С., Шарп А.Дж., Моллер Р.С., Ульманн Р., Капур Р., Пинкель Д., Купер Г.М., Вентура М., Роперс Х.Х., Томмеруп Н., Эйхлер Э.Е., Белланн-Шантелот К. Повторяющиеся реципрокные геномные перестройки 17q12 связаны с почечной недостаточностью. болезней, диабета и эпилепсии. Am J Hum Genet. 2007 ноябрь;81(5):1057-69. Epub 2007 г., 26 сентября. Цитирование в PubMed или бесплатная статья на PubMed Central .
- Нагамани СК, Эрез А, Шен Дж, Ли С, Родер Э, Кокс С, Каравити Л, Пирсон М,
Кан С.Х., Саху Т., Лалани С.Р., Станкевич П., Саттон В.Р., Чунг С.В. Клинический
спектр, связанный с повторяющимися геномными перестройками в хромосоме 17q12.
 Eur J Hum Genet. 2010 март; 18(3):278-84. дои: 10.1038/ejhg.2009.174. Epub 2009 Октябрь
21. Цитирование в PubMed или бесплатная статья в PubMed Central
Eur J Hum Genet. 2010 март; 18(3):278-84. дои: 10.1038/ejhg.2009.174. Epub 2009 Октябрь
21. Цитирование в PubMed или бесплатная статья в PubMed Central - Нагамани СК, Чжан Ф., Щелочков О.А., Би В., Оу З., Скалья Ф., Пробст Ф.Дж., Shinawi M, Eng C, Hunter JV, Sparagana S, Lagoe E, Fong CT, Pearson M, Doco-Fenzy M, Landais E, Mozelle M, Chinault AC, Patel A, Bacino CA, Sahoo T, Kang SH, Cheung SW, Lupski JR, Stankiewicz P. Микроделеции, включая YWHAE в Область синдрома Миллера-Дикера на хромосоме 17p13.3 приводит к дисморфизмы, ограничение роста и когнитивные нарушения. J Med Genet. 2009 г.Декабрь; 46 (12): 825-33. doi: 10.1136/jmg.2009.067637. Epub 2009, 6 июля. Цитирование в PubMed
- Потоцкий Л., Би В., Тредуэлл-Диринг Д., Карвалью К.М., Эйферт А., Фридман Э.М.,
Глейз Д., Крулл К., Ли Дж. А., Льюис Р. А., Мендоса-Лондоно Р., Роббинс-Фурман П., Шоу С.,
Ши X, Вайссенбергер Г., Уизерс М., Яценко С.А., Закай Э.Х., Станкевич П., Лупски
младший Характеристика синдрома Потоцкого-Лупски (dup(17)(p11.
 2p11.2)) и
определение дозозависимого критического интервала, который может свидетельствовать об аутизме
фенотип. Am J Hum Genet. 2007 апрель; 80 (4): 633-49. Epub 2007 Feb 26. Цитирование в PubMed или бесплатная статья на PubMed Central
2p11.2)) и
определение дозозависимого критического интервала, который может свидетельствовать об аутизме
фенотип. Am J Hum Genet. 2007 апрель; 80 (4): 633-49. Epub 2007 Feb 26. Цитирование в PubMed или бесплатная статья на PubMed Central - Potocki L, Chen KS, Park SS, Osterholm DE, Withers MA, Kimonis V, Summers AM, Мескино В.С., Аньяне-Йебоа К., Кашорк К.Д., Шаффер Л.Г., Лупски Дж.Р. молекулярный механизм дупликации 17p11.2 – гомологичная рекомбинация, обратная Микроделеция Смита-Магениса. Нат Жене. 2000 янв; 24(1):84-7. Цитата в PubMed
- Браузер генома UCSC: статистика
- Юань Б., Харел Т., Гу С., Лю П., Бурглен Л., Шанто-Бастаро С., Геловани В., Бек
CR, Carvalho CM, Cheung SW, Coe A, Malan V, Münnich A, Magoulas PL, Potocki L,
Лупски Дж.Р. Неповторяющиеся события перегруппировки 17p11.2p12, которые приводят к двум
Сопутствующие геномные нарушения: дупликация смежных генов PMP22-RAI1
Синдром. Am J Hum Genet. 2015 5 ноября; 97 (5): 691-707.
 дои:
10.1016/j.ajhg.2015.10.003. Цитирование в PubMed или бесплатная статья в PubMed Central
дои:
10.1016/j.ajhg.2015.10.003. Цитирование в PubMed или бесплатная статья в PubMed Central - Zody MC, Garber M, Adams DJ, Sharpe T, Harrow J, Lupski JR, Nicholson C,
Сирл С.М., Уилминг Л., Янг С.К., Абуэллей А., Аллен Н.Р., Би В., Блум Т., Боровски
ML, Bugalter BE, Butler J, Chang JL, Chen CK, Cook A, Corum B, Cuomo CA, de Jong
П.Дж., ДеКаприо Д., Дьюар К., Фитцджеральд М., Гилберт Дж., Гибсон Р., Гнерре С., Гольдштейн
С., Графхэм Д.В., Грокок Р., Хафез Н., Хагопиан Д.С., Харт Э., Норман Х.Х., Хамфри С.,
Джаффе Д.Б., Джонс М., Камаль М., Ходияр В.К., Лабутти К., Лэрд Г., Лехоцки Дж., Лю Х,
Lokyitsang T, Loveland J, Lui A, Macdonald P, Major JE, Matthews L, Mauceli E,
Маккэрролл С.А., Михалев А.Х., Мадж Дж., Нгуен С., Никол Р., О’Лири С.Б., Осоэгава К.,
Шварц Д.С., Шоу-Смит С., Станкевич П., Стюард С., Сварбрек Д., Венкатараман В.,
Уиттакер К.А., Ян Х., Циммер А.Р., Брэдли А., Хаббард Т., Биррен Б.В., Роджерс Дж.,
Ландер Э.С., Нусбаум К. Последовательность ДНК хромосомы 17 человека и анализ
перестройка в человеческом роду.
